MGAM mutations in melanoma
Right after I came up with the μ-wheel visualization, people immediately followed up with the obvious question: “looks nice, but what is next?”
If you, like I do, work in the field of Computational Biology, the chances are that you are already familiar with the following situation:
- Unbelievable amount of information is publicly available for analysis (e.g. TCGA).
- You pick one particular question, out of millions, and start crunching the data in a systematic manner to answer your question.
- You end up with a huge list of results, i.e. multiple answers to your question.
- You browse these results and see that some of them are so good to be true, some not-so-bad and some not interesting to you at all.
- And you start thinking about what to do with all these results.
I found myself clueless about what to do next (as step 6) and every time this happened, I started asking people around me for advice on what to do. It took me four years of graduate-level education and hours (sometimes even days) of discussions to realize that I, often, do not have many options but to take either of the following two paths:
- Cherry-pick one really interesting result and try to go for a validation, that is often accompanied with wet-lab experiments.
- Show that these results can be used, again in a systematic fashion, to predict a particular phenotype (e.g. survival time for each patient).
And I should mention that these paths are of interest to me only when I want to get some secondary yet publishable results. It is rather unfortunate that the primary results are often considered not to be sufficient for any type of publication, hence they frequently get lost in the transition to secondary results due to various things (negative results, time, etc.).
Let’s get back to the two possible paths and try to focus on the former. The problem with follow-up experiments is that you need money, time, instruments and, of course, expertise on the subject of interest. Computational Biology labs often lack one or more of these components, therefore have to look for collaborations, as it is supposed to be. And here comes the problem of convincing somebody that what you want to do is interesting – and this problem is a really really hard one.
How, in the world, you can convice your collaborator about doing experiments on that gene that nobody has ever heard of before but that also shines as a pearl in your result set? Well… based on my experience, majority of time, you cannot – and I find this really sad. Really sad that we are just trashing some random observations like nothing just because they do not look promising.
So that is why I am going to mention this gene, called MGAM, in this post as an example to this situation, although I know that this is going to be a quite far-reaching post.
MGAM, maltase-glucoamylase, mutations in melanoma
While eye-balling the μ-wheel, I realized that there is this gene called MGAM (maltase-glucoamylase) that have a few recurrent mutations across many cancer studies. So I went to cBioPortal, as usual, and submitted a cross-cancer query to see how all mutations in this gene look like. Below is a cross-cancer histogram showing relative frequencies of alteration (mutations) in MGAM:
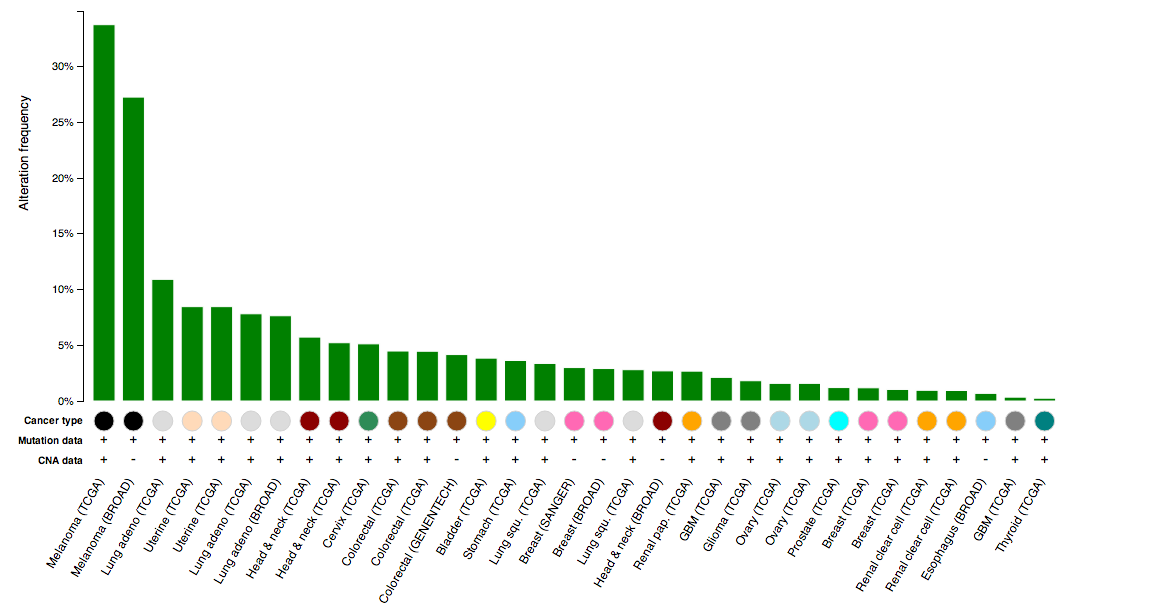
Notice that the two studies to the far left, the ones that have the highest alteration frequency, are all melanoma characterized by two different studies? This is interesting – possibly indicating they do something in a tissue-specific way. How about looking at how the mutations pile-up across all these studies:
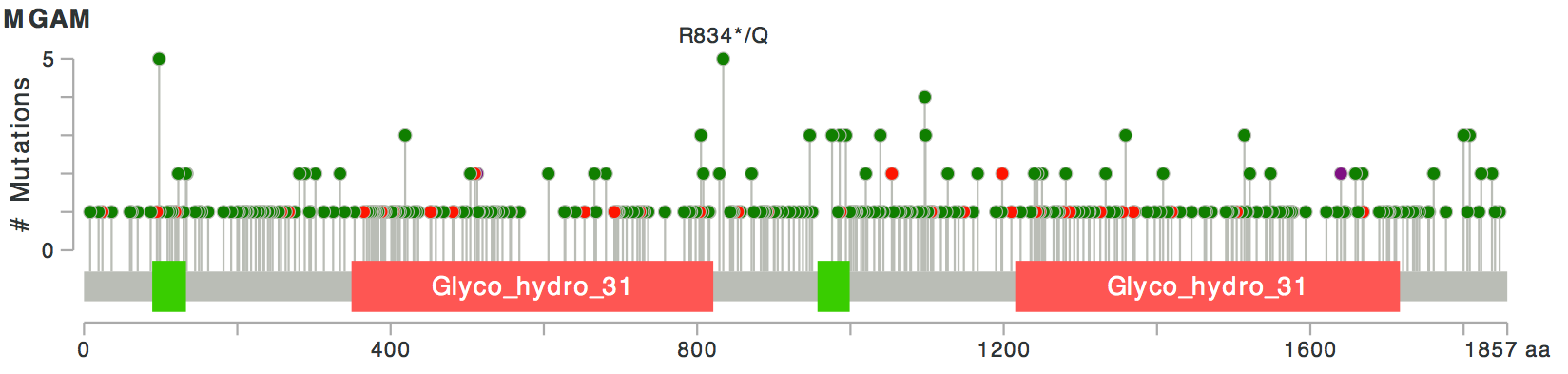
Let’s assume that this gene is not one of the TTN-like fishy genes. Then, looks like, as was also apparent from the wheel, there are a few recurrent mutations in this gene. Recurrent mutations, and the fact that the mutations have relatively high allele frequencies in mutated samples, are good signs: it means that these mutations possibly help the cells in some way, hence they were not selected against and did not get lost in the process.
Furthermore, most of these mutations seem to be missense mutations (green) and only a few of them, truncating mutations (red). This, although weakly, implies the missense mutations are possibly activating rather than disabling, because otherwise, we would expect to see relatively more truncating mutations.
If activating, then what do all those mutations affect the function of the protein? Considering that this gene is annotated as a maltase-glucoamylase (ECs: 3.2.1.20 and 3.2.1.3), we can assume that for MGAM-mutated cells, the reaction this enzyme catalyzes runs more effectively and faster – meaning that we have more breakdown of maltose-like species into glucose for cancer cells than it is for normal cells.
This is interesting because we do know that cancer cells require more glucose uptake to support their incredibly high proliferation rate, again compared to the normal cells. For the sake of the argument, let’s assume that the mutant protein causes more glucose availability hence is more advantageous to the cancer cells; but what can we do about it?
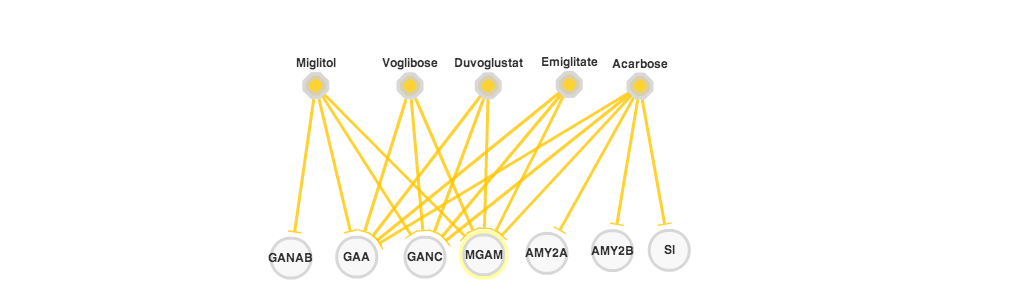
No worries, I have some good news for you: first, looking at the PiHelper data, looks like there are FDA-approved drugs (orange hexagons in the figure above) that inhibit the activitity of this gene and are already commercially available; second, MGAM is a membrane protein with an extracellular domain, making it easy to raise potentially blocking antibodies against it.
So, the over-reaching idea is as follows:
Then, the follow-up experiment to this hypothesis should be pretty straight-forward and relatively cheap:
- Obtain MGAM-mutated and MGAM-wildtype cell lines.
- Obtain all available drugs targeting MGAM.
- Test whether MGAM-mutated cell lines are more sensitive to inhbition with any of these drugs compared to MGAM-wildtype ones.
But finding a collaborator or funding for these experiments?… Well, I don’t think it is going to be that easy.