Of Synthetic Lethals and Vulnerabilities in Cancer
The Dream
Imagine a cancer patient walking into the clinic to learn her therapy options. Her tumor sample was collected some while ago and now it is time to hear the lab results. Results, of course, are from a comprehensive -omics analysis of the tumor sample and are tailored for her. As soon as they pop-up on the physician’s screen, the physician takes a careful look at them and prescribes a drug for the patient suggested by the analysis. The drug is no common cancer drug; it is a drug that has been on the market for some while but is being used for treating another disease. It turns out, from the -omics analysis of this patient, this drug should magically kill only the tumor cells and hence should not have any side effects for the patient. Patient starts on the therapy, and as expected, the tumor starts shrinking and eventually disappears. The patient goes on with her life normally during/after the therapy. She is happy to hear the good news and leaves the clinic with a smile. As she leaves, another cancer patient walks into the clinic to learn his personalized therapy options…
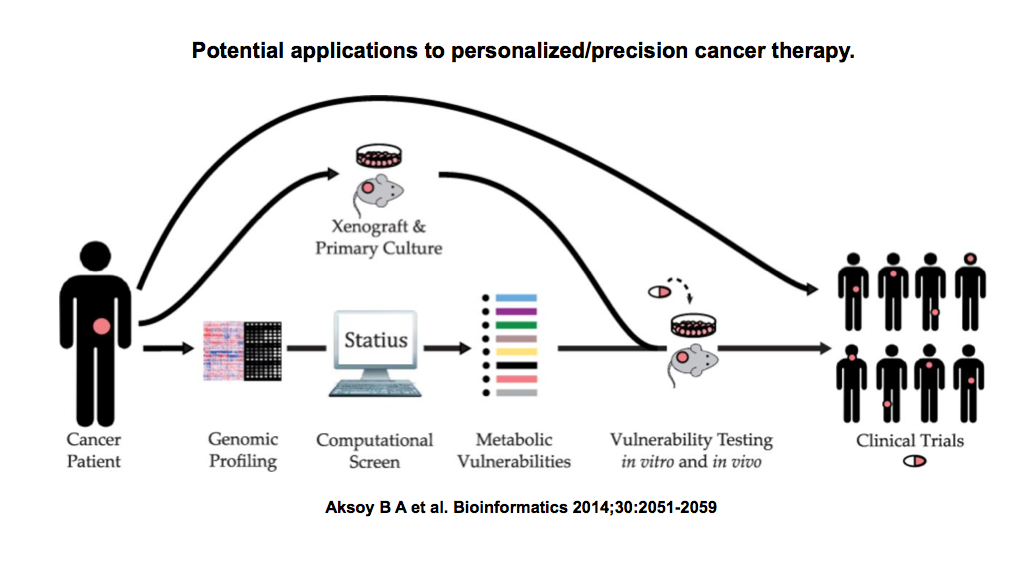
The above scenario, although it may sound as too good to be true, is not completely impossible with the latest technology and our current understanding of the cancer genome. In fact, many universities and instutions are already moving in this direction and started thinking seriously about the way they approach this personalized medicine idea. And in the meantime, the need for a computational tool or a set of tools that will give us some idea about which patient should be treated with which drug is becoming more apparent every single day.
Treating a tumor that has HER2-amplified with trastuzumab is a no-brainer. Nor it is for a BRAF V600E mutant patient with vemurafenib. But what if the patient doesn’t have any of those actionable alterations? How do you decide on the magical drug to use in the treatment? Computational approaches are to rescue!
The Concept
But before going into these computational approaches, let’s take a moment to step back and take a look at a few other concepts. Synthetic lethality, for example, is an important one. If gene A and B are synthetic lethal pairs, it simply means that cells can do fine without gene A or without gene B but not without both. Then there are essential genes. Essentiality is highly context-specific, meaning that a gene might be essential for cells when a specific condition is met. A trivial example to this are synthetic lethal pairs, where one of these genes become essential when the other one is knocked out. That is, gene A is essential when B is already knocked-out. Sometimes you can get more complex patterns, where, for example, gene C might become essential when gene A is over-active. It is also worth noting that these two terms can be, from time to time, be used interchangeably.
These concepts are even more interesting when we are talking about cancer, because cancer cells are a mess with their instable genomes. In the process of a normal cell to become a cancer cell, a cell accumulates lots of random alterations. And not surprisingly, these alterations sometimes disable one of the partners of a synthetic lethal pair. Or due to a gain-of-function alteration, these might cause some other genes to become essential for the cancer cell. Cells can tolerate these events considerably well until either the cell loses the partner in the synthetic lethal pair or we perturb this cell and hit the other partner, for example, by a targeted drug. The former, we don’t see much in the data sets, because those cells without its essential genes are selected against and immediately disappear from the tumor. The latter, it is interesting, because these are the events that create vulnerabilities in cancer cells. Since such vulnerabilities are restricted to the cancer cells, they also create an opportunity for us to exploit them as a therapeutic approach. You can think of this as cancer cells losing their gene A and then us hitting gene B with a drug, hence selectively killing these cancer cells. And be aware that, since gene A is lost only in cancer cells, normal cells in the body will still do fine in this case, i.e. the ideal therapy with minimum toxicity to the host.
The Approach
Now that we know what the problem (cancer) and even a possible solution to it (therapeutic vulnerability), we can start exploring the approaches to reveal clinically relevant synthetic lethal gene pairs.
Let’s start with a trivial example and assume the following: every gene pair represents a synthetic lethality (SL) group. Assuming that we know about 40000 gene symbols, this extreme approach leads to around 160 million potential SL pairs. This is just a way of suggesting SL pairs, but is it helpful? Not really so.
Instead, let’s be more realistic and work with in vitro models: cell lines. We can, for example, take a bunch of cell lines for which we know the genetic alterations, and try to hit every single gene they have one by one and observe the effects. The genes, when hit, that kill some of the cell lines are interesting, because they are potentially essential to those cells and represent therapeutic targets. This is not a bad idea at all and in fact, there are really nice studies (Project Achilles and Marcotte et al.) out there that conduct this type of a screen on cell lines. The technology of choice for these experiments is usually pooled-shRNAs combined with deep-sequencing. You can also swap shRNAs with drugs and get a similar data set centered around compounds (e.g. CCLE, CancerRxGene or CTRP). The problem with this type of an experimental approach, however, is that it is really hard to scale things up, because these experiments are expensive and labor-intensive. And cell line pools do not always represent the full patient diversity, hence are under-powered.
Yet another way to approach this problem is to model cells, that is working computationally on a systems-level. You can, for example, learn how normal and cancer cells operate on a metabolism level and observe what happens to these cells if you hit a gene in both. The ideal hit disrupts the metabolism in cancer cells bad enough to kill them or stop them proliferate, but does not effect the normal cells. This is a feasible and reasonable approach (see Folger et al.), but for this, you need to rely on generalized normal/cancer models that cover a majority of the patients but not all of them.
Or you can leverage what we know about metabolism so far in a qualitative manner and assume that cells need all these reactions to function correctly. Then you can take a look at all enzymes that catalyze the same reaction in a cell, so-called isoenzymes, and see if a cancer cell lacks one particular isoenzyme, where the other partner can be targeted with a drug. The idea here is that losing a metabolic enzyme creates a vulnerability for the cancer cells, and you can exploit this vulnerability by perturbing these cells with a drug to inhibit an isoenzyme partner. We recently implemented this method and report our findings here. The advantage of this approach is that it allows to nominate vulnerabilities and drugs associated with them in a personalized manner. The disadvantage is that you rely on prior knowledge and not all isoenzymes that you infer from knowledge bases might represent SL gene groups.
Finally, you can simply ignore the modeling and the prior knowledge parts, and let the data speak for itself. Given the amount of public data that we have, an integrated analysis will tell us which genes are potentially SL pairs –by looking at their co-alteration frequency and expression patterns– and whether there is experimental data, e.g. shRNA screen results, to back this observation up. As this really nice study by Jerby-Arnon et al. shows, using these data is no easy task and requires combining many smart methods for proper integration and nomination. But it seems doable and at the end provides us with a general network of all SL pairs.
The Conclusion
No matter what kind of method or approach we adopt, accumulating results from all these synthetic lethals and essentials are no doubt exciting. With the increasing amount of data and new smart methods, we will soon have our huge catalog of therapy options with specific contexts that they are expected to work. And once we reach to that completeness, we will be much closer to the level where we treat cancer as if it is a chronic disease. And I am looking forward to those day while trying to contribute to this ultimate aim as much as I can.
The Rant
For those that are curious, here is me talking about our approach and results during the 3rd TCGA Scientific Symposium.